


A partir de la teoría microbiana de Pasteur, los científicos comenzaron a comprender los mecanismos que gobiernan la producción de las enfermedades transmisibles, y de este modo el Hombre se preparó —en lo mucho o poco que estuviera a su alcance— para defenderse.
Los agentes responsables de las enfermedades infecciosas pasaron a ser, entonces, clasificados entre virus, bacterias, hongos, algas y protozoos. De entre ellos, los primeros no se cuentan entre los seres vivos.
Ello es así porque entre las características que definen la vida se cuentan, primero, poseer los dos tipos de ácidos nucleicos (ADN y ARN) y, en segundo lugar, que el organismo en cuestión sea capaz de sintetizar sus propios suministros, que disponga de una maquinaria química operable y que sea capaz de autorreplicarse o reproducirse.
Bacterias, algas, hongos y protistas son por supuesto capaces de todo ello, pero los virus no. Ellos tienen sólo un ácido nucleico (nunca los dos). Esta diferencia ha permitido clasificarlos en dos grandes grupos: los virus (que poseen ADN) y los retrovirus (que sólo tienen ARN).
El virus, además, no posee los medios químicos para producir nada: ni su propia energía, ni su propio alimento, nada en absoluto. Se trata de una simple cápsula de glucoproteínas que esconde en su interior unas moléculas de ácido nucleico. El virus se adosa a una célula viva, le inyecta su ADN (o ARN) y el mismo toma el control de la maquinaria química de la célula. La víctima, entonces, abandona sus operaciones vitales normales y dedica toda su energía y capacidad, durante lo que le quede de vida, a fabricar... nuevos virus. Cuando la célula está repleta de virus "hijos", sencillamente estalla y muere, liberándolos para que puedan infectar a otras infortunadas células normales.
La lucha entre el Hombre y la enfermedad se centró entonces, desde hace tiempo, en los virus y los microbios patógenos.
Hasta ahora.
A principios de la década de 1900, una extraña y mortífera enfermedad comenzó a ganar prensa en los medios científicos. Parecía afectar solamente a la etnia Fore, individuos pertenecientes a comunidades geográficamente aisladas de la zona sur de Nueva Guinea, en aquel entonces bajo jurisdicción australiana, y en el idioma del lugar se conoce como kuru.
Comenzaba con una amplia y gravísima serie de manifestaciones neurológicas que parecían ser de naturaleza degenerativa. Gadjusek, uno de los investigadores que mejor la estudiaron, descubrió en el kuru tres fases o etapas, solamente referidos a la evolución de los síntomas nerviosos. El primer nivel o fase ambulatoria presenta inestabilidad de la bipedestación, la marcha, los movimientos de las manos, la voz y los movimientos oculares y la mirada. El discurso verbal comienza a deteriorarse, aparecen temblores y escalofríos incontrolables, falla la coordinación neuromuscular en las extremidades inferiores que comienzan a moverse lentamente hacia arriba, y un trastorno oclusivo del habla —una especie de atragantamiento— que se conoce como disartia. El segundo nivel de progresión de los síntomas, llamado fase sedentaria —porque el paciente ya no puede caminar sin ayuda— incluye temblores más severos, ataxia (incoordinación muscular), convulsiones musculares y una gran labilidad emocional característica, en la que el enfermo pasa de la euforia a la depresión, se ríe a carcajadas, a los cinco minutos llora desconsoladamente, y así sucesivamente. A pesar de todo ello, la degeneración muscular está ausente, y todos los músculos esqueléticos presentan todavía sus reflejos normales.
La tercera fase de la enfermedad, o fase terminal, se caracteriza por un agravamiento de todos los síntomas: el paciente no puede permanecer sentado sin ayuda, la disartia y los temblores empeoran. Aparece la disfagia (imposibilidad de tragar), incontinencia fecal y urinaria y profundas ulceraciones de la piel. Luego, inevitablemente, viene la muerte entre horribles convulsiones.
 Miembro de la etnia Fore enfermo de kuru |
Como se ve, todos los síntomas hacen pensar en una enfermedad neural degenerativa, y muy rápida. Gadjusek describe los tiempos de la evolución del kuru: desde la aparición de los primeros síntomas hasta la muerte transcurren entre tres y seis meses (tres meses en la inmensa mayoría de los casos).
Durante las décadas de 1920 y 1930, los científicos occidentales se rompieron la cabeza estudiando al kuru, mientras veían morir a ingentes cantidades de la población indígena del sur de Nueva Guinea. La epidemia —que de eso se trataba— fue creciendo descontroladamente, hasta alcanzar su pico máximo en los años ´60.
¿Qué causaba esta espantosa enfermedad?
Para 1950, el estado australiano había identificado perfectamente el foco de la enfermedad, que pasó a ser llamado Fore del Sur. Gadjusek estuvo en Nueva Guinea desde 1957 hasta 1996 estudiando esta devastadora patología.
Los médicos anteriores habían observado que el kuru parecía tener preferencia por las mujeres: de hecho, en los once años que mediaron entre 1957 y 1968, 1.100 de los 8.000 habitantes indígenas de Fore del Sur murieron de kuru (14% de la población; en
un país como la Argentina eso hubiese significado más de 5 millones de cadáveres). De las 1100 víctimas, murieron ocho veces más mujeres que hombres. A partir de los años ´60, se observó un catastrófico avance del kuru también entre los niños y los ancianos.
Como normalmente las víctimas caían en grupos familiares, no faltaron quienes postularon que el kuru era una enfermedad congénita y hereditaria, provocada por un gen mutante heredado de padres a hijos. Esto no podía ser, y la explicación es muy sencilla:
un gen tan agresivo, que ataca a las víctimas a tan temprana edad y con una letalidad tan grande (100% de mortalidad), muy pronto quedaría fuera del pool genético de la población afectada. Simplemente, las víctimas no tenían tiempo de reproducirse para
transmitir el gen patológico a la descendencia.
Gadjusek se dio cuenta de esto y en 1966 comenzó a inyectar a chimpancés con material orgánico de las víctimas del kuru (sangre, orina, materia fecal, tejido muscular y nervioso). Pronto los animales desarrollaron también la enfermedad. La verdad era clara: el kuru era una enfermedad infecciosa, transmisible entre los seres humanos y también entre especies diferentes.
El mecanismo de transmisión del kuru era especialmente horrible: los indígenas de Nueva Guinea eran caníbales. Practicaban una suerte de ritual fúnebre antropofágico hacia sus seres queridos, y ello era el motivo de que los brotes de la enfermedad se produjeran entre individuos pertenecientes a una misma familia. La monstruosa incidencia entre las mujeres se debía a que ellas eran las encargadas tradicionales de desmembrar el cadáver del pariente y de prepararlo para su ingestión ritual. Al morir un ser querido, las mujeres quitaban al cadáver los brazos y los pies, separaban la carne de los huesos, quitaban los sesos del cráneo y abrían el tórax y el abdomen para retirar los órganos internos. Luego, repartían entre los familiares la carne (y especialmente el cerebro, con el que preparaban una especie de sopa) como alimento. Otro médico que investigó el kuru en los años 60 y 70, Lindenbaum, afirma que los muertos por causa del kuru eran más apreciados para comer en estas fiestas religiosas, porque su grasa corporal tenía el sabor de la del cerdo, siempre y cuando la víctima hubiese muerto rápidamente —lo cual, como hemos dicho, era casi siempre el caso—. Esta grasa de "cerdo largo" muerto de kuru era repartida entre las mujeres que manipulaban el cuerpo, sus niños pequeños y sus padres y ancianos. Aquí tenemos la apariencia "hereditaria" perfectamente explicada.
Al haber demostrado la transmisibilidad de la enfermedad al chimpancé, Gadjusek pensó que se trataba de un virus. Lo llamó "virus lento" por los aparentemente largos períodos de incubación y latencia que presentaba el kuru, de entre unos dos y 23 años. Sin embargo, las enfermedades virales producen indefectiblemente una respuesta del sistema inmunitario y pueden encontrarse anticuerpos para ese virus. Nada de esto ocurría con el kuru. Parecía una enfermedad virósica, pero lo cierto era que el organismo del paciente y en particular su respuesta inmune —o más bien la falta de ella— estaban totalmente divorciadas de la dinámica orgánica del virus. Poco tiempo después, Gadjusek tuvo que darse por vencido. Si bien había comprobado que el kuru era infeccioso, menester era aceptar la falsedad de la teoría viral. El agente productor del kuru no era un virus.
Mientras Lindenbaum y Gadjusek se devanaban los sesos tratando de entender la dinámica del kuru, comenzaron a aparecer enfermedades similares —al menos en cuanto signos y síntomas— en diversas especies animales y también en el ser humano. No podía tratarse de kuru, pues ya se sabía que éste era exclusivo de los neoguineanos caníbales, pero los síntomas, el pronóstico y la evolución de aquellas enfermedades eran por cierto sospechosamente parecidos a los del kuru.
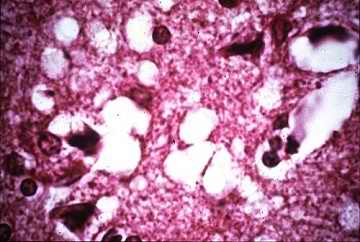 Destrucción cerebral en una víctima del kuru. Obsérvense los orificios, vacuolas y canales dejados por las neuronas ausentes |
Así, los veterinarios comenzaron a estudiar enfermedades como el tembleque de cabras y ovejas, la encefalopatía transmisible del visón, el síndrome de cansancio crónico de las mulas, la encefalopatía espongiforme felina, el síndrome de Creutzfeldt-Jakob (SCJ), la encefalopatía de los ungulados exóticos, el síndrome de German-Straussler-Scheinker (SGSS), el síndrome de Alpers (SA) y el insomnio familiar fatal (IFF). Poco tiempo después, todos estos males comenzaron a conocerse en conjunto como "encefalopatías espongiformes" (EE), porque el cerebro de sus pacientes, en examen post-mortem, se presentaba taladrado de pequeños túneles, con pérdida de masa encefálica, y se parecían a esponjas.
Fue entonces cuando llegó el "mal de la vaca loca".
Ya los estudios en Nueva Guinea con monos habían demostrado que este tipo de encefalitis eran transmisibles de una especie a otra. Sin embargo, no todas pasaban al hombre. El ser humano había demostrado ser susceptible a cuatro de ellas: el Creutzfeldt-Jakob, el German-Straussler-Scheinker, el insomnio familiar fatal y el kuru. Jamás se había visto un ser humano con cuadros similares al del tembleque, la encefalopatía del visón, la del gato ni la de los ungulados exóticos.
Sin embargo, las vacas comenzaron a enfermar en Europa. Esto era extraño, porque la vaca es un ungulado pero los síntomas de los pacientes no se parecían a los de los ungulados exóticos: se asemejaban más a los del tembleque de las cabras y ovejas. Las vacas comenzaron a presentar signos alarmantes, y a morir en cantidades cada vez más altas...
¿Podría el consumo humano de carne bovina transmitir esta extraña encefalopatía espongiforme al hombre como si del canibalismo de los indios se tratase? Y de ser así, ¿cómo lo había contraído la vaca inglesa? Las vacas no comen cabras, ya se sabe...
En 1993, una mujer se presentó en el Hospital General de Massachusetts quejándose de una fuerte depresión. Sus síntomas incluían hipoactividad, pérdida de la memoria de corto plazo e incontinencia urinaria. Tenía sólo 47 años, por lo que era demasiado joven como para padecer de una de las enfermedades degenerativas del sistema nervioso del anciano como el mal de Alzheimer. En pocos meses comenzó a perder estabilidad y a sentir mareos. La tomografía axial computada mostró una ligera atrofia cerebral y central. Tres años más tarde, las respuestas de la mujer a cualquier pregunta eran totalmente inadecuadas, reía como loca sin motivo, lloraba al instante siguiente y temblaba mucho. Una segunda TAC mostró el mismo problema en el cerebro. La atrofia central comenzó a avanzar, siempre acompañada por el ciclo depresión-euforia, los temblores y la inestabilidad. ¿Era kuru? ¿En medio de Boston? Su discurso se volvió incomprensible, la atrofia cerebral siguió avanzando, y la desdichada mujer murió en el Hospital General entre convulsiones, con apenas 53 años de edad.
No era kuru: se acababa de describir el primer caso de síndrome de Creutzfeldt-Jakob, el "mal del humano loco".
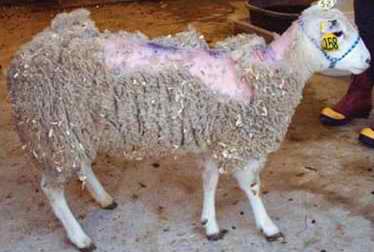 Oveja enferma de tembleque. La pérdida de lana es consecuencia de las úlceras dérmicas. En Gran Bretaña se alimentaba a las vacas con estos animales |
Los granjeros ingleses habían comenzado a alimentar a sus vacas con tripas y menudos de ovejas y cabras, merced a los altos costos del alimento balanceado común. De esta forma, el tembleque de las ovejas y las cabras había pasado al ganado bovino y se había transformado en una enfermedad nueva: la encefalitis espongiforme bovina (EEB) o "mal de la vaca loca", que no es más que la versión vacuna del tembleque. Al ingerir carne u otros tejidos infectados, la EEB pasa al humano. Así como ella es la forma bovina de la enfermedad de las ovejas, el SCJ es la versión humana de la EEB. El desastre estaba en camino: sólo hacía falta comenzar a exportar animales enfermos para que se convirtiera en una catástrofe. Una enfermedad similar al kuru —sólo que más lenta en llegar al desenlace— estaba a punto de arrojarse sobre los países centrales.
 Lamentable aspecto de una vaca con encefalopatía espongiforme ("mal de la vaca loca") |
Sin embargo, los científicos aún no tenían en claro qué tipo de agente patógeno producía las encefalitis espongiformes. Imposible, entonces, encontrar el remedio para una enfermedad producida por un agente desconocido.
La falta de explicación de la naturaleza exacta del kuru había desesperado a los estudiosos durante más de medio siglo. Pero la respuesta estaba disponible desde 1982.
Ese año, Prusiner había identificado y definido a las encefalitis espongiformes como enfermedades "priónicas". Su definición de prión fue "una partícula infecciosa compuesta por una molécula de proteína". ¿De qué hablaba Prusiner? ¿No sabíamos desde siempre —y aquí llegamos a la lista de nuestro primer párrafo— que las enfermedades infecciosas eran producidas por algas, hongos, virus, bacterias y protozoos?
En efecto, Prusiner tenía razón. El kuru, la EEB, el SCJ, el SGSS, el IFF y las encefalitis de los ungulados, del gato y del visón son enfermedades a priones. Una simple molécula proteica que ni siquiera posee ADN, que no es un virus ni una bacteria y que, por consiguiente, no es identificada por el sistema inmune como un agente agresivo al que hay que eliminar. Otras versiones de la misma proteína son normales en el organismo, y en este hecho estriba la explicación de que las encefalitis espongiformes no presentan evidencia de respuesta inmune ni producción de anticuerpos. Los priones son proteínas propias, "nuestras"... pero producen enfermedades espantosas, todas ellas mortales en el 100% de los casos.
Cohen demostró, en 1994, que los priones causan la amplia variedad de enfermedades neurológicas que hemos enumerado. Las EE pueden ser infecciosas, hereditarias o esporádicas. La causa de las EE esporádicas se desconoce. Las EE hereditarias se producen porque el prión es producido por la mutación de un gen que debiese haber producido la proteína normal. El gen priónico se transmite a la descendencia, como en el caso del IFF, EE priónica hereditaria por excelencia. La tercera forma de la enfermedad es la infecciosa, que se transmite por contacto o consumo alimentario de tejidos enfermos, de la misma o de diferente especie.
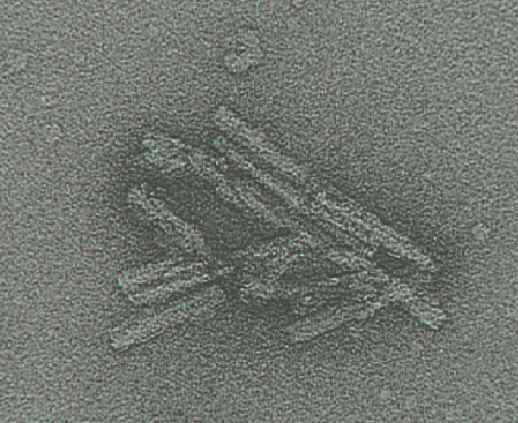 El prión al natural: microfotografía de un cerebro humano infectado de Creutzfeldt-Jacob |
Todas las EE son producidas por la mutación de un único gen, que en el humano se denomina PrP. Cuando es normal, produce una proteína llamada PrPC (la "C" por "celular", ya que está presente en todas las células del organismo). Existen al menos veinte mutaciones patológicas en el hombre del gen PrP. En lugar de producir la proteían PrPC, producen otra diferente llamada PrPSc ("Sc" por "scrapie", nombre inglés del tembleque ovino). La mutación se produce en un solo punto del gen (concretamente el codón 102).
Prusiner estudió en 1995 a la proteína PrPC, la comparó con la patológica PrPSc, y descubrió que en la secuencia de los 15 aminoácidos terminales de la misma, una molécula del aminoácido lisina había sido sustituida por prolina en la proteína anormal. Esta simple mutación de un solo aminoácido determina que toda la gigantesca estructura molecular de la proteína se vea alterada.
El cambio de lisina por prolina hace que un octapéptido (un grupo de tan sólo 8 aminoácidos) de un extremo de la proteína comience a replicarse, a copiarse indefinidamente, yendo cada nueva copia a modificar a su vez a otra molécula de PrPC normal para
convertirla en PrPSc. De este modo se explica la replicación del prión (que no es, recuérdese, una partícula viva en sí misma y ni siquiera un virus). Es una proteína deforme que produce "mensajeros" que, a su vez, deforman a nuevas moléculas normales.
La forma normal de la proteína está formada por alfa hélices que rodean una columna central espiralada. La PrPSc, en cambio, tiene fibras tipo beta colocadas alrededor de una columna central recta y extendida. ¡Y todo por el cambio de un solo aminoácido!
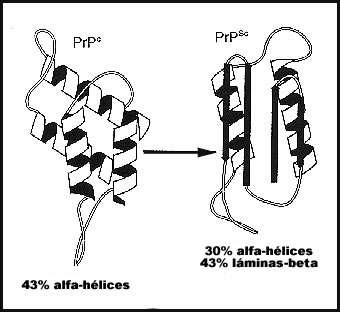 Cambio de estructura entre la versión normal y la deforme |
Como el lector imaginará, los científicos pronto comprendieron que el estudio minucioso de la proteína priónica, su replicación y el resto de su dinámica era fundamental para la comprensión de las enfermedades a priones, especialmente la EEB y su sucedáneo primate, el SCJ.
La enorme diferencia de estructura entre las versiones normal y anormal de la proteína del gen PrP demuestra que hay cambios postraducción, además del cambio de un aminoácido. Se llama traducción al proceso mediante el cual se interpreta el contenido del gen y se lo traduce como estructura proteica. Esta estructura está malformada en las EE, y sigue cambiando después de la traducción.
Sin embargo, algunas teorías manifestaban que los priones no eran solamente la molécula proteica: algunos afirmaban que debía tener también un trozo de ADN o de ARN, lo que lo asemejaría mucho más a un virus.
Miles de laboratorios en todo el mundo se dedicaron a tratar de probar o descartar esta teoría: lo que hicieron fue tomar tejidos infectados de EE y someterlos a fuertes bombardeos de rayos ultravioleta duros y radiaciones ionizantes, que destruyen los ácidos nucleicos. Luego, inyectaron estos tejidos a sus animales experimentales. Todos desarrollaron la enfermedad. Ello significaba que no había ADN ni ARN en el prión. Quedaba por probar el aserto contrario, a saber, que se trataba de la molécula proteica sola y aislada. Sometieron entonces tejido cerebral de ovejas con tembleque a tratamientos desproteinizadores, como detergentes fuertes que desnaturalizan las proteínas. Los priones fueron inactivados. Se trataba de proteínas solas.
Un nuevo agente infeccioso acababa de descubrirse. Un nuevo enemigo acechaba en nuestros genes, en nuestros animales, en nuestra propia comida y Dios sabía en donde más.
El asunto de ubicar un método que permita luchar contra el prión no es simple. Las diferencias estructurales entre la PrPC y la PrPSc hacen que esta última sea mucho más resistente que la original. La forma normal se disuelve en detergentes no desnaturalizantes y es muy sensible a la acción de las proteasas, enzimas cuya función es destruir proteínas. En cambio, la versión mutante no se disuelve y es parcialmente resistente a las proteasas. La PrPSc, además, es totalmente estable en medios fuertemente ácidos y alcalinos (es decir, sobrevive en medios de pHs de entre 2 y 10) y sobrevive, emergiendo perfectamente entera e infecciosa, luego de un baño de dos años sumergida en formol.
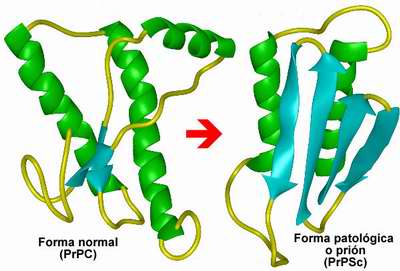 Diagramas tridimensionales de la proteína normal y su derivado priónico |
El prión tampoco se desnaturaliza ante las altas temperaturas.
La horrible naturaleza del prión se vuelve, entonces, evidente. La cocción de los alimentos nada puede contra el prión. Los detergentes normales, tampoco. Ni el ácido clorhídrico (pH 2) ni la lavandina concentrada (pH 9-10) logran hacerle mella. El formol no la afecta.
La única solución ante un tejido infectado con priones es la incineración sumaria y efectiva, convertirlo en cenizas, por ejemplo, en un horno crematorio.
Es la única medida segura y razonable.
La conversión de una proteína normal en un prión ocurre de la siguiente manera: cuando una molécula de PrPSc entra en contacto con una de PrPC, la "pliega" o "rota", reemplazando el aminoácido necesario y obligándola a adoptar la estructura, forma o "posición" de un prión. El prión no se destruye en el proceso, por lo que puede seguir "prionizando" moléculas normales. Los priones recién reclutados van de inmediato a "convertir" a más moléculas normales. Se trata de una verdadera e inimaginable reacción en cadena de mutaciones, donde una proteína normal es atacada por una célula enferma, enfermada a su vez, y dotada de la nueva y espantosa capacidad de infectar a toda molécula sana que, desde ese momento, se cruce en su camino.
Las EE son enfermedades del sistema nervioso central porque las células que mejor albergan y soportan este proceso son, precisamente, las neuronas. Los priones se acumulan en los lisosomas de las células nerviosas, convierten a millones de proteínas normales en nuevos priones, y el lisosoma estalla liberando a sus nuevos proselitistas. La neurona finalmente estalla también, soltando a los priones en el medio intercelular para seguir cumpliendo su miserable misión. Los huecos dejados por las células muertas forman los canalículos que dan a los cerebros enfermos su característico y letal aspecto "espongiforme".
Todas las células nerviosas poseen un receptor que es "palpado" por las proteínas "asesinas" que son encargadas de destruir las células viejas o anormales. La célula sana se salva de la muerte porque una proteína "tapa" o bloquea este receptor de la membrana, de manera que la proteína asesina no pueda encontrarlo. Es como poner masilla en la cerradura para que no se pueda introducir la llave. Como el lector seguramente sospecha, la encargada de bloquear el receptor en la célula sana es la versión normal de la PrPC. La versión priónica no puede llenar el receptor, de modo que las proteínas asesinas se confunden y eliminan neuronas sanas pensando que son anormales. Comienzan por una y siguen por las que están conectadas a ellas, dejando "túneles" espongiformes. He aquí la sencilla pero letal dinámica interna de las EE.
Pasemos ahora a un breve análisis de cada una de las EE humanas (ya hemos hablado del kuru):
El SCJ infeccioso, como hemos dicho, proviene de la transmisión del prión de otra especie al ser humano, principalmente por ingestión. La forma hereditaria se transmite de padres a hijos, porque el gen PrP mutante produce el prión en vez de la proteína normal. No se conocen las causas de la forma esporádica, que sólo tiene una prevalencia de un caso por millón de habitantes por año.
Por cada 100 casos de SCJ, se reportan 2 de SGSS. Se estima que 1 de cada 10.000 personas son portadoras de este síndrome al momento de la muerte. Eso implica que hay unos 4.000 enfermos de síndrome de German-Straussler-Scheinker en la Argentina al momento de escribir este artículo. Sin embargo, se sabe que estas cifras están grandemente infravaloradas, porque muchos de los casos de ECJ y EGSS son víctimas de errores de diagnóstico al ser confundidos con el Mal de Alzheimer u otras enfermedades neurológicas degenerativas. Ambas enfermedades provocan pérdida del control muscular, desgano, parálisis y la muerte, típicamente después de una grave neumonía. El análisis post-mortem muestra un sistema nervioso central traspasado de canales, con grandes vacuolas de espacio vacío, depósito de fibras amiloides y astrogliosis.
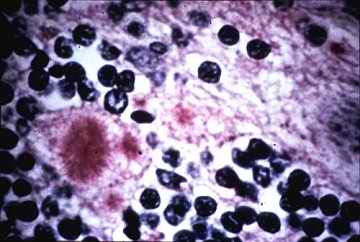 Los daños provocados en un cerebro humano |
La EGSS se presenta entre los 40 y 50 años, con ataxia cerebelosa, problemas motores de suma gravedad, poca incidencia de demencia y cursa durante varios años hasta conducir a la muerte. La ECJ, por su parte, incluye demencia y los pacientes difícilmente sobreviven un año. Ambas patologías se creían solamente esporádicas, pero ahora se sabe que son también hereditarias y/o infecciosas.
El IFF, mucho más común en España que en los demás países, se caracteriza, como su nombre lo indica, por un insomnio intratable que es consecuencia de una atrofia espongiforme del tálamo. También conduce a la muerte sin remedio.
El Síndrome de Alpers se define como cualquiera de estas enfermedades a priones en lactantes o niños.
La transmisión de las enfermedades priónicas en animales se han estudiado principalmente en vacas y cabras. Se han descubierto solamente dos formas de transmisión. Una de ellas es la hereditaria.
La otra vía es la ingesta. La mala costumbre de permitir a los animales parir en los mismos terrenos donde otros pastan hace que queden residuos de amnios o placenta infectados en el pasto, dando lugar de este modo a que el prión se transfiera de un animal enfermo a uno sano. La aberrante técnica inglesa de dar a las vacas tripas de oveja como alimento determinó también el explosivo avance de la enfermedad.
En el hombre, aparte de la transmisión hereditaria, hay varias formas de contagiarse el prión. La ingesta de carne o vísceras infectadas es la principal, pero se han demostrado casos en que la mala esterilización de los instrumentos quirúrgicos ha producido la enfermedad. La administración de hormona de crecimiento de origen bovino se sabe que ha transmitido la EEB a seres humanos, y el transplante de una córnea de un muerto de ECJ o EGSS también las provoca en el inocente y desprevenido receptor. Otros mecanismos comprobados de transmisión del prión son: electrodos estereotáxicox, endodoncias, transplantes de tímpano, de duramadre e inyección de hormonas sexuales, especialmente gonadotrofinas.
Sin embargo, no todo está perdido. En 1995, Prusiner comenzó a trabajar en ratones de laboratorio, que fueron sometidos a una terapia que destruía el gen PrP. De esta manera, el científico descubridor del prión produjo ratones libres del gen. Según sus trabajos publicados, los ratones no mostraron ningún efecto en absoluto ni anormalidad alguna. Así que es posible que el PrP no sea un gen esencial. Si esto se demuestra, con una terapia de antígenos "anti.PrP" podría llegar la solución para estas catastróficas enfermedades. Gracias a sus trabajos, Prusiner recibió el Premio Nobel de Medicina en 1997.
 Fibras amiloides depositadas en el cerebro de un niño francés muerto de Síndrome de Creutzfeldt-Jacob. El prión le fue contagiado por el médico al inyectarle somatotrofina (hormona de crecimiento) infectada |
Atacando el problema desde otro ángulo, Serpell estudió el problema de las fibras amiloides. Una fibra amiloide es el resultado de hacer pasar a una proteína soluble por un determinado proceso que la deposita en forma de fibra lineal, recta, altamente estable llamada "amiloide". Esto es lo que sucede con la PrPSc, y por ello es tan resistente a la ruptura. Serpell no llama a estas enfermedades EE ni "priónicas" sino "amiloidosis" por este motivo.
Las fibras amiloides tienen ciertas características especiales: no se ramifican y su superficie es suave, sin anfractuosidades; se tiñen con rojo Congo y exhiben un patrón de refracción de los rayos X muy determinado. Comprender el proceso íntimo de la producción de fibras amiloides y los modos de detenerlo también podría ayudar en la terapia antipriónica.
Mientras la gente se infecta y muere en todo el mundo (especialmente en el ámbito anglosajón por los motivos apuntados), la Organización Mundial de la Salud ha dictado pautas de bioseguridad a cumplir para tratar de frenar el avance de la enfermedad. Ellas son:
Por otra parte, la OMS ha definido los métodos de esterilización que sirven para destruir el prión en tejidos u objetos infectados:
Ningún otro método sirve para la esterilización, como no sea la cremación en horno.
Así, no surten ningún efecto los sistemas siguientes:
Todos aquellos que trabajen con materiales sospechosamente priónicos deben utilizar máscara, anteojos de seguridad, doble par de guantes y delantal plástico. Todos los elementos que han estado en contacto con los materiales sospechosos deben ser sumergidos en ácido fórmico durante una hora, luego lavados con formol al 40% y por último esterilizados en una de las formas descriptas.
Otras recomendaciones incluyen el uso en humanos de hormonas exclusivamente obtenidas por ingeniería genética, y de productos provenientes de países libres de EEB (vaca loca). Como veremos, existe un solo país en estas condiciones, por lo que "libres de" debe leerse "con esporádicos casos de".
Increíblemente, la leche, la gelatina, el sebo y los derivados de estos materiales, aún si provienen de animales infectados, se consideran seguros.
Como se ha visto, la EEB se descubrió en el Reino Unido en 1986. Sólo dos años más tarde, cuando los demás países, incluida la Unión Europea estaban aún en veremos al respecto, la Argentina estaba ya implementando su propia estrategia para prevenir la aparición de priones animales en su ganado de consumo humano.
En efecto, en 1988 mi país formó un comité compuesto por técnicos del CICV-INTA (Instituto Nacional de Tecnología Agropecuaria), SENASA (Servicio Nacional de Sanidad Animal) y la UNLP (Universidad Nacional de La Plata), todos ellos organismos oficiales. A ellos sumó especialistas del sector privado argentino (SERONO). Todos ellos interactuaron con y fueron capacitados por especialistas internacionales: en el Reino Unido (CVL-Weybridge), los Estados Unidos (NHI y The Scripp Research Institute) e Italia (Universidad de Roma).
La Argentina no tenía un solo caso reportado de vaca loca ni de tembleque de la oveja cuando se desató la enfermedad, y por lo tanto todas las medidas fueron destinadas a impedir el ingreso de priones al territorio nacional. Argentina prohibió de inmediato (1990) la importación de semen, óvulos y embriones, así como de productos bovinos, ovinos y caprinos, del Reino Unido de Gran Bretaña hacia la Argentina (Resolución 429/90 de la Secretaría de Agricultura, Ganadería y Pesca). Esta norma continúa en vigencia. Se instauró el análisis permanente de los factores de riesgo de priones. La Argentina estaba libre de ellos, por lo que el único factor de riesgo era la importación desde Inglaterra o países infectados.
 Incineración de vacas enfermas en Inglaterra |
En 1992, la Argentina obtuvo la validación de sus esfuerzos efectuados y proyectos a futuro en París. Al año siguiente, Argentina comenzó el análisis de muestras de cerebro de animales muertos que fuesen sospechosos de portar enfermedades neurológicas o que hubiesen llegado al matadero con síntomas de enfermedad. 1.200 muestras de otros tantos bovinos fueron analizadas, y ninguna de ellas manifestó signos de priones o de encefalitis espongiforme.
En 1995, por Resolución 252/95 del SENASA, la Argentina prohibió el uso de carnes y huesos de oveja para alimentación de rumiantes (algo que a ningún ganadero argentino se le ocurriría ni en sus más perversas pesadillas). Igualmente, ahora está prohibido.
La Argentina practica el seguimiento de los animales importados previamente a la aparición de la enfermedad. Los ganaderos argentinos que importaron semen, óvulos, embriones o animales en pie de Inglaterra antes de 1986 están sujetos a la inspección y
seguimiento del SENASA hasta la muerte por sacrificio o natural de los animales implicados, y sus restos no pueden ser utilizados en ninguna forma, aunque no den muestras de estar infectados, y deben ser destruidos por incineración.
Todas estas excelentes acciones preventivas y los estudios científicos realizados por la Argentina cumplen en un todo con las condiciones y recomendaciones de la Organización Mundial de la Salud y la Organización Internacional de las Epizootias, convirtiendo a la Argentina hoy (2005) en el primer y único país del mundo total y certificadamente libre de Encefalitis Espongiforme Bovina o Mal de la Vaca Loca.
NOTA FINAL DEL AUTOR: Con dolor y vergüenza, debo reconocer que no todo el territorio argentino está libre de EEB. Las Islas Malvinas, pertenecientes a la Argentina y usurpadas por Gran Bretaña en el siglo XIX, tienen una de las mayores prevalencias mundiales de Mal de la Vaca Loca, en razón de haber recibido exportaciones de carne y ganado en pie desde Inglaterra.